16th August 2024
NovaLead licenses its Patented Repurposed Drug to Ipca Laboratories. The drug is launched in India with a brand name Diulcus®.
- NovaLead and Ipca have announced the licensing deal for marketing, sales and distribution of Diulcus® in India market.
- The launch of Diulcus® marks a pivotal moment for the people suffering from DFU.
- Media Link: Business Standard
NovaLead is now actively exploring to out-license the product in other territories. Please contact us for more details
NovaLead is a drug discovery research company focused on discovering new indications for generic drugs. Enabled by proprietary technology platform, NovaLead's drug repositioning approach is targeted at finding new biological targets for generic drugs, thereby discovering new indication possibilities. This approach is distinctly differentiated from conventional line extension strategies. NovaLead's business development and outsourced research services are managed by its wholly owned subsidiary - Novalead Pharma Inc., USA.
NovaLead's discovery pipeline has an addressable market estimated at over US$ 10 billion p.a. The lead candidate in the pipeline is Galnobax®, which is a topical gel for the treatment of hard to heal Diabetic Foot Ulcer (DFU). The recently concluded Phase – III clinical trial has demonstrated that Galnobax® has successfull met both primary and secondary endpoints for wound closure and is safe for long term use. Given the limitations of existing therapies for DFU, Galnobax® is looked at as a ray of hope by millions of DFU patients worldwide.
Entire discovery research of NovaLead is done in-house and entire biological validation is outsourced to service providers with specific expertise around the world. The commercial model of NovaLead is to outlicense / partner its discoveries after first human proof of concept. Headquartered in Pune - India, NovaLead is funded by top tier venture investors from India and Europe.
Management Team

Supreet Deshpande
Managing Director
Supreet is a founder and visionary behind NovaLead. With an international career of over 23 years in various senior strategic positions... know more

Atul Aslekar
CTO
Atul is CTO and co-founder of NovaLead. Atul has over 22 years of experience in technology and operations management... know more

Dr. Sudhir Kulkarni
VP, Discovery
Sudhir has been a backbone of scientific developments at NovaLead. He has been proponent of drug repurposing for past 20 years... know more
RESEARCH FOCUS and PIPELINE
NovaLead’s research effort is focussed on find new thepaeutics for hard-to-treat diseases and unmet medical needs. NovaLead explores generic drugs to find new indication possibilities in these areas. Such candidates have little chance of failure on account of safety as their safety profile in humans is well documented through historic use for their original indication.Thus eliminating one of the major causes of late stage drug failures. NovaLead's' pipeline has several candidates at different stages of development. The lead compounds in NovaLead's pipeline are given below:

Galnobax®
Galnobax® is a topical gel for non healing chronic wounds. Galnobax® is a repurposed generic drug molecule, which has recently completed Phase- III clinical trial in diabetic foot ulcer (DFU) successfully meeting both primary and secondary endpoints for both, safety and efficacy. The data from Phase III and Phase I/II studies substantiates pre-clinical findings that Galnobax® re-triggers natural wound healing processes impaired due to diabetic condition.
Global DFU challenge
World over, more than 392 million people have Diabetes, expected to rise to 592 million in 2035. About 15% of them suffer from DFU at least once in lifetime, with 25% of DFUs eventually requiring amputation. DFU is a leading cause of lower extremety amputation. Every 20 seconds somewhere in the world, a lower limb is amputated due to DFU. Five year mortality post amputation is 46%. DFU remains a serious disease with very limited drug options and very high cost of treatment.
DFU imposes substantial burden on public and private payers, in the range of US $9-$13 billion in US alone. Due to rising number of diabetics globally, the DFU treatment market is expected to cross US $ 11 Billion by 2027.
Globally 392 million people have
Diabetes; expected to rise to 592 million in 2035
About 15% of diabetics suffer
from DFU at least once in lifetime
About 85% of non traumatic lower extremity
amputations are due to DFU

NLP91
NovaLead has received grant of its patent for NLP91 covering broad set of indications including psoriasis in USA. NovaLead team has discovered novel anti-inflammatory activity for an FDA approved generic drug molecule having potential for inflammatory skin diseases like psoriasis and dermatitis. The discovery of the indication was achieved by extensive use of in-silico RVHTS technologies. A series of experimental validations by in vitro studies also confirmed the activity of the candidate for psoriasis. The candidate NLP91 is under further pre-clinical validations in animal models.
Market for Psoriasis / limitations of existing drugs
Psoriasis is a chronic skin disease that 2-3 % of the global population. It is the most prevalent autoimmune disease in the U.S. with more than 7.5 million Americans affected (~ 2.5% of the population). The disease negatively impacts quality of life for majority of patients. Broad level Psorisis and related market is expected to reach $60.5 Billion in 2032.
Many biologics approved for arthritis are in use for treating severe posiriasis. There are fewer drugs for treating mild-to-moderate psoriasis, which constitute more than 70% of psoriasis cases. Due to limitations of these drugs, there is a dire need for an effective and longer lasting treatment for mild-to-moderate psoriasis.
Psoriasis affects about 125
million people worldwide
As of 2015, annual medical costs for psoriasis are estimated
at USD 3.9 billion
Psoriasis has been linked to the depression and suicidal tendencies
in patients
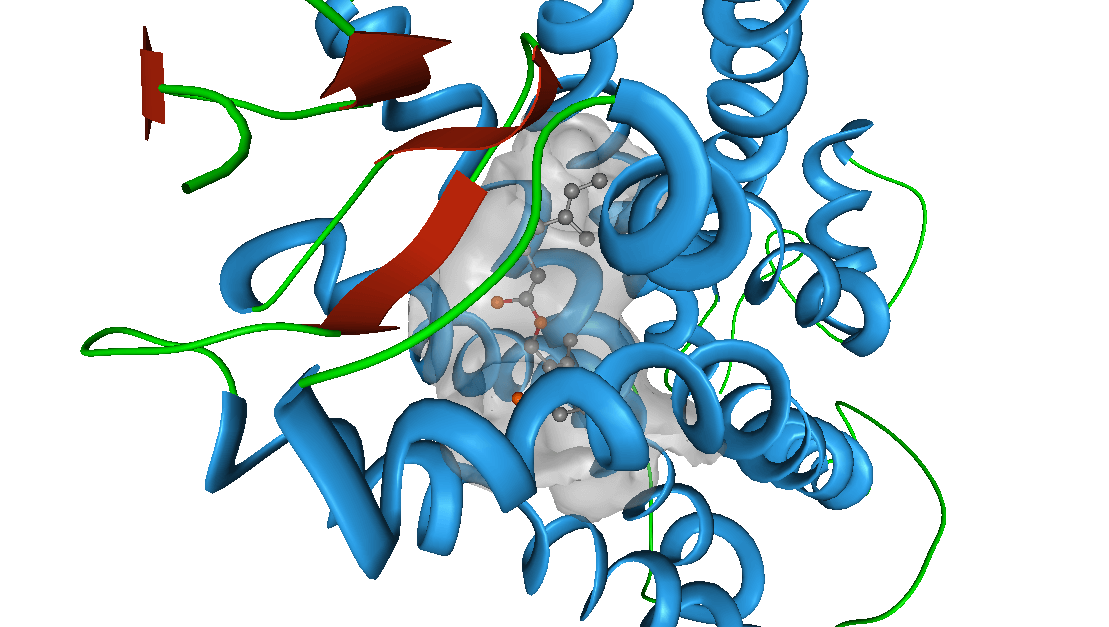
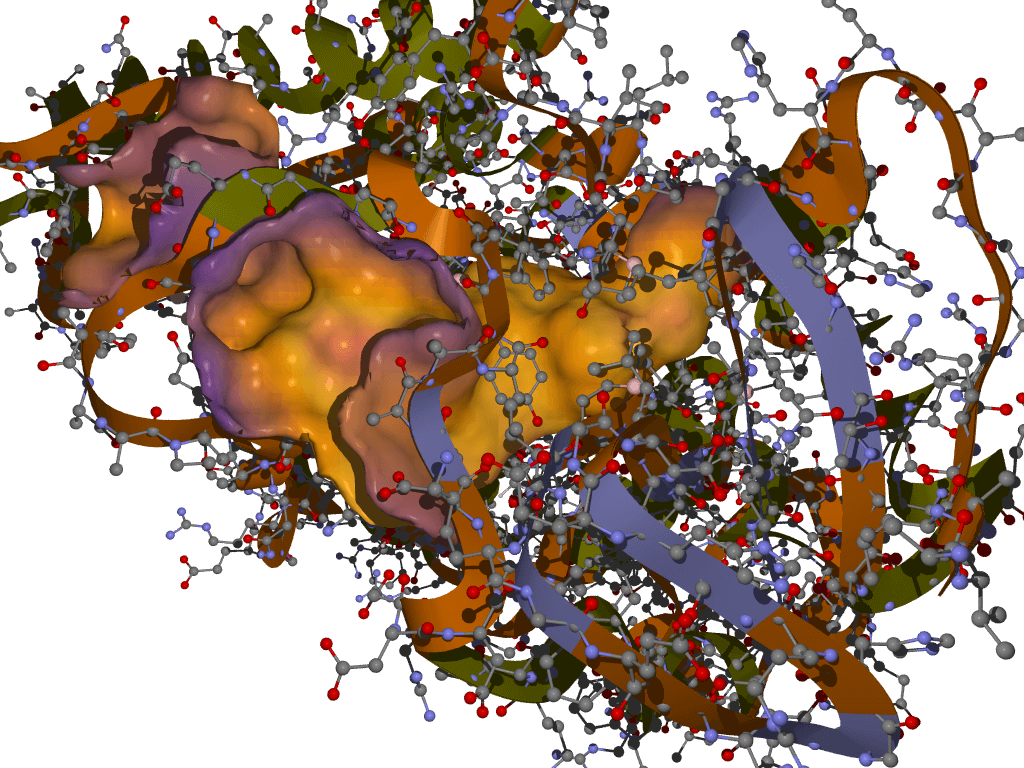
RVHTS TECHNOLOGY
The Reverse Virtual High Throughput Screening (RVHTS) blends structure guided and ligand based drug design and screening approaches. It includes methods for flexible receptor-drug docking interaction analysis, molecular shape based similarity identification, molecular fragments to activity relationship studies and active site structure analysis to name a few. Besides this, it employs extensive drug-disease-target knowledgebase which is acompendium of databases of FDA approved drugs, validated drug targets, mechanism of actions for various diseases and the intelligent correlations between these datasets. The platform set up consists of modern multi-core parallel processing cluster which churns millions of molecular simulations to deliver an all inclusive interaction analysis.
Application
NovaLead's' RVHTS technology can be effectively employed for product life cycle management and reviving shelved compounds. It also assists in identifying most appropriate disease for an early stage molecule. NovaLead team either starts with the intended drug molecule or preferred protein target or the disease of interest. If this interests you, please visit us on www.vlifesciences.com
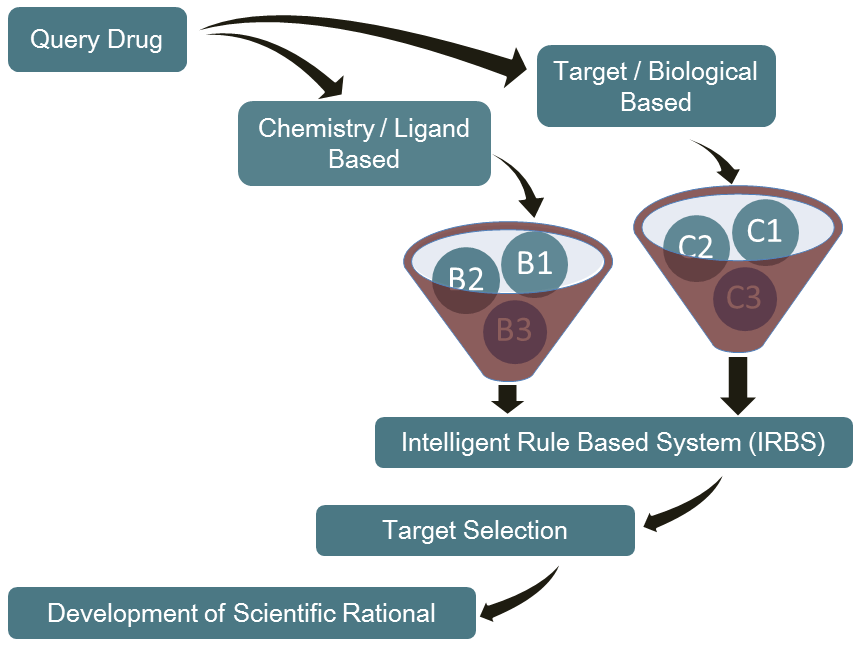
PARTNERING
NovaLead believes in the spirit of partnership and collaboration. We are open to explore partnering as well as licensing alternatives. We expect our drug candidates to follow the 505(b)(2) route for FDA filing and similar regulatory paths for EU, Japan and other regulated markets.
Beside the product candidates in our pipeline, we are also open for customer-driven collaboration. We can help you find novel indications of existing drugs, using our proprietary discovery platform.
To know more drop in an e-mail
or give us a call

NEWS and EVENTS
Galnobax®receives market authorization from CDSCO
24th April 2024: NovaLead's Patented Repurposed Drug Galnobax® receives market authorization from CDSCO for the treatment of Diabetic Foot Ulcer (DFU). The approval is based on the Phase 3 clinical trial data which established NovaLead’s drug Galnobax® promotes the complete closure of DFU significantly better than the Standard of Care ,which is the present treatment of choice for DFU. With this approval, Company will be able to manufacture and distribute the drug in Indian market through its sales and marketing partner. Galnobax® is among the few patented repurposed drugs which is completely developed in India, from discovery to market authorization.
Galnobax® data gets published in JAMA Open Network
15 May, 2023: The Phase 3 Clinical Trial data of Galnobax® got published in the reputed International journal, JAMA Open Network. The title of the research paper is - Topical Esmolol Hydrochloride as a Novel Treatment Modality for Diabetic Foot Ulcers: A Phase 3 Randomized Clinical Trial.
Read the paper here
The Phase 1/2 clinical trial data of Galnobax® was published in the well known International journal, Advances in Wound Care. Read the paper here
Details of the pre-clinical studies and their data for Galnobax® were published in the reputed International journal, Frontiers Endocrinol. Read the paper here
Galnobax® achieves Primary Endpoint in the Phase-3 clinical trial for the treatment of Diabetic Foot Ulcers (DFU)
01 Dec 2021: NovaLead has recently completed Phase-III clinical study of Galnobax® which is its flagship product which is a repurposed drug. The Phase-III trial data analysis shows that the primary as well as secondary endpoint for percentage of subjects achieving complete ulcer closure are met with Galnobax demonstrating statistically significant superiority over the standard of care (SoC). Analysis further showed that Galnobax® demonstrated statistically significant superiority over the standard of care (SoC) in ‘Hard to Heal’ ulcers as well. The safety analysis showed that Galnobax® is safe for long term use.
NovaLead Pharma Pvt. Ltd. announces commencement of double blinded, multicenter Phase-3 study of Galnobax® for Diabetic Foot Ulcer
12 Nov 2018: NovaLead Pharma Pvt. Ltd. announces commencement of double blinded, multicenter Phase-3 study of Galnobax® for Diabetic Foot Ulcer, which is a global unmet medical need. Earlier clinical trials conducted by the company demonstrated promising efficacy and safety of Galnobax® in treating diabetic foot ulcers. Galnobax® is a repurposed generic drug molecule with a long safe history of human use. This Phase-3 human trial being conducted on 350 patients of DFU is partly supported by Grant-In-Aid by the Government of India. The results from trial are expected to be used to support registration and market approval in India and other developing markets.
NovaLead receives "Frost & Sullivan's Indian Drug Repurposing Technology Innovation Leadership Award 2017"
Novalead Pharma has been selected by Frost & Sullivan, leading global industry tracking firm as Best Technology Company in Drug Repositioning for the Year 2017. The award was received by our CEO Mr. Supreet Deshpande in presence of distinguished business and industry leaders in a glittering function held on 05 Oct 2017 at ITC Maratha, Mumbai. Previously in 2015, NovaLead Pharma also received the Best Innovation Company BIRAC award from DBT. Mr. Deshpande’s interview conducted by Frost & Sullivan will be shared in coming days.


NovaLead receives "DBT-BIRAC Innovator Award 2015"
NovaLead team is pleased to inform that the company has been selected for the “BIRAC Innovator Award 2015” from DBT, Govt. of India in recognition of the significant contribution made towards the high level of innovation research for “Determining safety, effective dose and frequency of application of Galnobax®”.
The selection has been made by an eminent Jury set up by the Secretary, DBT & Chairman, BIRAC and the award will be conferred during the Inaugural session of “4th BIRAC Innovators Meet” to be held on 15th – 16th September, 2015, at Heritage Village Resort, Manesar, Gurgaon”.
On this occasion Mr. Supreet Deshpande, CEO said:
"We are delighted at being selected for BIRAC Innovation Award - 2015 in healthcare. This award underlines the national relevance of our investigational drug product Galnobax® for diabetic foot ulcer. It is also a validation for NovaLead's innovative research approach to find new therapeutic products for unmet and hard to treat diseases. I congratulate the entire NovaLead team on this momentous occasion."
CAREERS
NovaLead is an equal opportunity employer with a stimulating work culture that fosters creativity and innovation. At NovaLead, the vision of contributing to a healthy, happy and convenient life for humanity is reflected in the working environment. We firmly believe that freedom of initiative and thinking are the fundamental rights of creative minds to excel in what they do and in what we stand for as a company.
Send your Resumes to careers@novaleadpharma.com

CONTACT US
NovaLead Pharma Pvt. Ltd.
2nd and 3rd Floor, Plot No-05,
Next to Sapling Nursery, Ram Indu Park,
Survey No-131/1b/2/11, Baner Road,
Pune 411045, Maharashtra.
Tel : + 91 7066233033
+ 91 8669656660
Enquiries
NovaLead Pharma Inc.
433 South Street
Shrewsbury MA 05415

NovaLead Pharma Inc.
NovaLead Pharma Inc. is a wholly owned subsidiary of NovaLead Pharma Private Limited, Pune – India (Indian parent company). It is located at 433, South Street, Shrewsbury MA 05415.
Its primary business activities are to manage conduct of:
-
Phase II B clinical trials for parent company’s discovery, Galnobax® which is a repurposed drug for Diabetic Foot Ulcer. The right to develop this discovery for USA is with Novalead Pharma Inc. The actual clinical trial conduct will be outsourced to third party CRO in USA.
-
Preclinical studies and subsequently, clinical Phase I/II trial for NLP-91 for Psoriasis in USA. The patent for this discovery is already filed by Novalead Pharma Inc. The actual clinical trial conduct will be outsourced to third party CROs in USA and research labs preferably within MA.
-
To manage preclinical studies for other candidates in discovery pipeline of the parent company in USA. The actual biological validation will be outsourced to third party research labs preferably within MA.
Galnobax®:
India Phase III has commenced in Nov 2018 and the Global Phase II-B is to commence soon.
Advance
NLP91 for Psoriasis to IND
Manage NovaLead collabration in USA
-
Patent filings for the Company and its Indian parent company’s discoveries across the world in consultation with Patent attorneys based in USA from time to time. Currently, the company works with Crowell and Moring LLP, Washington DC.
-
Oversee company’s collaborative research programs with government research institutions and universities in North America.
Supreet Deshpande
Managing Director
Supreet is the visionary behind NovaLead. With an international career of over 23 years in various senior strategic positions in large companies like Mahindra & Mahindra Ltd and Bajaj Auto Ltd, Supreet is a business development person at heart. Developing new markets, designing and implementing business strategies and leading market linked product strategies are some of the key areas of his contribution. With business development experience in multiple geographies like the USA, Europe, Latin America, Africa and Asia, Supreet brings with him significant learning for building an efficient and result oriented organization.In 2011, Supreet recieved prestigious BioSpectrum Asia Entrepreneur of the Year 2011 Award. This year under Supreet's leadership NovaLead received DBT-BIRAC Best Innovator award in healthcare.
Atul Aslekar
CTO
Atul is CTO and co-founder of NovaLead. Atul has over 22 years of experience in technology and operations management. Previously he has worked with large international organizations like Fujitsu and Indian software enterprises like ZenSar. With experience of working in Japan, UK and USA, Atul brings an international outlook to the organization. At NovaLead, Atul has been a significant contributor in the evolution of its technology platform and in management of the Galnobax clinical trial.
Dr. Sudhir Kulkarni
VP, Discovery
Sudhir has been a backbone of scientific developments at NovaLead. He has been proponent of drug repurposing for past 20 years. He is Ph.D. in Theoretical and computational Chemistry from University of Pune. He did his post doctoral studies at Nagoya University, Japan. He has been faculty at Department of Chemistry, University of Pune for about 7 years. He worked as Senior Consultant at Mahindra British Telecom (Tech Mahindra) before joining VLife Sciences Technologies right from the inception of the Company. He has played pivotal role in the discovery, prelinical, formulation development as well as clinical development of Galnobax. He is currently focussing on development of new pipeline products. He has 60 research publications and 6 international patents to his credit.